Qui di seguito trovate una trascrizione libera del mio intervento al convegno “Aritmologia in Liguria – Core curriculum per lo specialista clinico”, tenutosi a Genova lo scorso 5 novembre 2022. E’ stato un evento densissimo di contenuti ed una straordinaria occasione di confronto tra clinici (e non solo), al quale è stato un onore poter prendere parte.
Il tema della compatibilità tra dispositivi impiantabili (e, in particolare, i dispositivi cardiaci impiantabili attivi) e risonanza magnetica è un argomento di grande delicatezza ed interesse per le implicazioni in materia di responsabilità per gli operatori coinvolti (radiologo ed aritmologo in particolare). Nel dettaglio, tre sono gli ambiti di rilievo:
- norme applicabili e linee guida
- regole in tema di responsabilità per lavoro d’équipe
- indicazioni per l’utilizzo off-label (cioè fuori protocollo) dei dispositivi.
Le linee guida in materia
La legge n. 24/2017 (cd. legge Gelli-Bianco) dispone, all’art. 5, che
“1. Gli esercenti le professioni sanitarie, nell’esecuzione delle prestazioni sanitarie… si attengono, salve le specificità del caso concreto, alle raccomandazioni previste dalle linee guida pubblicate ai sensi del co. 3 (…). In mancanza delle suddette raccomandazioni, gli esercenti le professioni sanitarie si attengono alle buone pratiche clinico-assistenziali (…).
3. Le linee guida e gli aggiornamenti delle stesse elaborati dai soggetti di cui al co. 1 sono integrati nel Sistema nazionale per le linee guida (SNLG) (…) L’Istituto superiore di sanità pubblica nel proprio sito internet le linee guida e gli aggiornamenti delle stesse indicati dal SNLG (…)”.
Le linee guida in materia sono essenziali perché, per quanto non costituiscano norme di legge, rappresentano il consenso del mondo scientifico su di un dato argomento in un determinato momento storico, cioè il cd. stato dell’arte, al quale viene parametrata la condotta dell’operatore sanitario.
La conformità della condotta dell’operatore sanitario alle linee guida riconosciute (purché rispondenti alle necessità e caratteristiche del caso concreto trattato dal sanitario) costituisce causa di non punibilità sotto il profilo penale qualora il medico, nello svolgimento della sua attività, pur avendo correttamente individuato le linee guida applicabili al caso concreto, causi il decesso o lesioni al paziente per imperizia nella fase esecutiva (art. Art. 590-sexies Codice Penale).
Attualmente, nel sito internet del Sistema nazionale per le linee guida (https://snlg.iss.it/), non è stata ancora pubblicata alcuna linea guida in materia.
Il riferimento sono dunque le linee guida internazionali: solo per citare le più recenti, ricordiamo
· Heart Rhythm Society, Expert Consensus 2017
· Consensus Statement on MRIs in patients with CIEDs, ACR (American College of Radiology) Manual on MR Safety, 2020
· ESC (European Society of Cardiology) Guidelines on cardiac pacing and cardiac resynchronization therapy 2021.
Normativa di riferimento
Un set di norme d’interesse per la materia in questione è contenuto del Decreto del Ministero della Salute del 14 gennaio 2021 (Determinazione degli standard di sicurezza e impiego per le apparecchiature a risonanza magnetica e individuazione di altre tipologie di apparecchiature a risonanza magnetica settoriali non soggette ad autorizzazione), in vigore dal 15 aprile 2021.
Questo decreto contiene norme in materia di sicurezza degli operatori che operano sulle apparecchiature a risonanza magnetica e dei pazienti che vengono a contatto con le stesse, le quali tuttavia finiscono, di fatto, per avere un impatto sulla responsabilità degli operatori sanitari.
In passato, l’analogo D.M. 2 agosto 1991 disponeva che
«debbono essere esclusi da analisi RM persone portatrici di pacemaker cardiaco; altre protesi dotate di circuiti elettronici; preparati metallici intracranici o comunque posizionati in prossimità di strutture anatomiche vitali; clips vascolari o schegge in materiale ferromagnetico»,
e ciò indipendentemente dal fatto che, sin dal 2008, fossero stati introdotti sul mercato dispositivi cardiaci impiantabili cd. “MRI conditional”, e cioè compatibili con l’utilizzo delle apparecchiature a risonanza magnetica, a condizione del soddisfacimento delle condizioni d’utilizzo indicate dal fabbricante.
Nel 2018 la situazione cambia, e anche il D.M. 14 gennaio 2021 ora conferma che
«L’entrata nella ZONA AD ACCESSO CONTROLLATO deve essere regolamentata garantendo: la valutazione dei rischi connessi per soggetti portatori di dispositivi cardiaci impiantabili attivi o altri dispositivi medici attivi o passivi (…) L’accesso al SITO RM di persone portatrici di dispositivi impiantati o di altri materiali o preparati dovrà essere valutato con la massima attenzione caso per caso».
Viene dunque meno il divieto radicale di sottoporre a risonanza magnetica i pazienti portatori di dispositivi attivi: ora l’indicazione di condotta è quella di valutare caso per caso la compatibilità della sottoposizione a risonanza magnetica con il dispositivo impiantato al paziente.
Per quanto concerne i pazienti portatori di dispositivi cardiaci impiantabili attivi, il D.M. 14.1.2021 fa inoltre obbligo alla struttura sanitaria di predisporre un modello organizzativo specifico, a garanzia della sicurezza della prestazione e della salute del paziente, che comprenda un processo di valutazione del rapporto rischio/beneficio dell’esecuzione/mancata esecuzione dell’ESAME RM, sotto la diretta responsabilità del medico responsabile della sicurezza clinica e dell’efficacia diagnostica dell’apparecchiatura RM (punto D.2 Criticità dell’ESAME RM).
Le responsabilità dei componenti dell’équipe
In punto, il DM 14.1.2021 dispone che:
– il medico responsabile della sicurezza clinica e dell’efficacia diagnostica dell’apparecchiatura RM deve tenere conto delle competenze professionali e delle necessità operative dei medici specialisti non di area radiologica che possono usufruire, per la loro attività, dell’ESAME RM e prevedere il coinvolgimento del cardiologo nell’esecuzione di prestazioni diagnostiche su pazienti portatori di dispositivi cardiaci impiantabili (punto E. RESPONSABILI)
– le richieste di esami devono essere vagliate dal medico responsabile della prestazione clinica (e cioè dal radiologo che sarà presente in loco al momento dell’esame). Questi, in base alla propria esperienza clinica, alla valutazione delle condizioni del paziente ed alla effettiva utilità dell’esame, deciderà sull’opportunità di accoglimento della richiesta e sulle modalità di esecuzione dell’esame stesso (D.4 Misure di sicurezza per i pazienti).
È dunque compito e responsabilità del radiologo di stabilire, sulla base dell’assenza di documentate controindicazioni del paziente, se effettuare o meno la risonanza magnetica.
Questa precisazione vale anche e soprattutto quando la richiesta di esame provenga da altri professionisti sanitari, ed in particolare dal medico di medicina generale.
In punto, la Cassazione ha recentemente precisato che
“Occorre, dunque affermare il principio per cui, in tema di colpa professionale medica, qualora ricorra l’ipotesi di cooperazione multidisciplinare, ancorché non svolta contestualmente, allorquando venga prescritto un esame diagnostico invasivo…,
Il medico specialista chiamato ad effettuare l’esame non può esimersi dal valutare, oltre che la presenza di fattori che possano condizionare negativamente l’esame stesso (assunzione di farmaci, parametri vitali, esito esami ematochimici), anche la bontà della scelta diagnostica operata dal medico richiedente in relazione alla sintomatologia lamentata dal paziente ed all’esistenza o meno di precedenti indagini diagnostiche che avvalorino il sospetto della malattia ipotizzata… Rispetto a tale valutazione… lo specialista non può lavarsene le mani, soprattutto quando la prescrizione dell’esame proviene da un collega che specialista non è.”
(Cass. Pen., Sez. IV, n. 30051 del 29 luglio 2022, che trovate commentata qui).
Anamnesi e consenso informato
In punto di anamnesi, il D.M. 14.1.2021 dispone che
“Ferme restando le competenze previste dalla normativa vigente per i diversi operatori sanitari coinvolti nell’esecuzione dell’esame, il paziente, prima dell’esecuzione dell’ESAME RM, è tenuto a rispondere alle domande contenute nel questionario che l’equipe RM utilizzerà per far emergere possibili controindicazioni all’esecuzione dell’ESAME RM. Il MEDICO RESPONSABILE DELLA PRESTAZIONE DIAGNOSTICA valuterà – sulla base delle informazioni acquisite – l’eventuale necessità di ulteriori approfondimenti per i quali dovrà essere garantita la possibilità di esecuzione di una visita medica atta allo scopo…
Il questionario anamnestico concernente le informazioni relative al paziente – da utilizzare secondo quanto definito in appendice 1 – deve prevedere i quesiti allo stato dell’arte delle conoscenze relativi alle possibili controindicazioni all’esecuzione dell’ESAME RM, e va predisposto ed eventualmente integrato sulla base delle scelte che competono al MEDICO RESPONSABILE DELLA SICUREZZA CLINICA E DELL’EFFICACIA DIAGNOSTICA DELL’APPARECCHIATURA RM.
Il questionario anamnestico deve essere firmato dal MEDICO RESPONSABILE DELLA PRESTAZIONE DIAGNOSTICA, mentre il paziente controfirma in calce alla medesima pagina, a testimonianza della propria consapevolezza sul possibile rischio connesso ad eventuali risposte false o mendaci ai quesiti sottopostigli…» (Punto B.6 Locale visita medica).
La sottolineatura della necessità di quesiti allo stato dell’arte accende una luce sulla necessità di periodico aggiornamento dei “modelli organizzativi specifici” che ciascuna struttura che intende installare un’apparecchiatura a risonanza magnetica è obbligata ad adottare al fine di renderli conformi alle linee guida più aggiornate.
In tema di consenso informato, il D.M. 14.1.2021 ricorda inoltre che
“Prima di effettuare l’ESAME RM il paziente deve essere informato sulle possibili controindicazioni, i rischi e le limitazioni di carattere medico.”
La precisazione, per quanto apparentemente ovvia, diviene ancor più essenziale in caso di utilizzo off-label (cioè fuori protocollo) dei dispositivi (es. sottoposizione a risonanza magnetica di pazienti portatori di dispositivi impiantabili MRI unsafe, cioè convenzionali, oppure di pazienti portatori di dispositivi MRI conditional, ma a condizioni diverse rispetto a quelle indicate dal fabbricante). Sul punto torneremo di seguito.
Il rapporto ISTISAN 15/9
Altro documento tuttora molto importante è il Rapporto ISTISAN 15/9 (titolato “Dispositivi cardiaci impiantabili attivi e risonanza magnetica: aspetti tecnologici, inquadramento normativo e modelli organizzativi”).
Questo documento, elaborato nel 2015 da un team di esperti coordinati dall’Istituto Superiore di Sanità,
«è stato redatto per fornire un supporto all’attività clinica di cardiologi e radiologi coinvolti nella pianificazione ed esecuzione di esami di risonanza magnetica in pazienti portatori di pacemaker, defibrillatori impiantabili e loop recorder, al fine di garantire la sicurezza del paziente e degli operatori. Gli autori ritengono che l’argomento oggetto del documento richieda una trattazione organica degli aspetti scientifici e tecnologici, normativi e organizzativi, relativi all’interazione tra i dispositivi cardiaci impiantabili attivi e la risonanza magnetica.»
Anche il Rapporto ISTISAN conferma l’essenzialità del coordinamento interdisciplinare tra il radiologo, medico responsabile della prestazione diagnostica, con le altre professionalità coinvolte nella prestazione diagnostica:
«Il radiologo, in qualità di medico responsabile della prestazione diagnostica, ai fini dell’applicazione del principio di giustificazione, ovvero della decisione ultima sulla sostenibilità dell’esame, si coordina con il cardiologo con competenza in elettrostimolazione, con il medico radiologo responsabile dell’attività dell’impianto, con il fisico medico esperto responsabile della sicurezza, con l’ingegnere clinico, ciascuno per le proprie competenze, acquisendo la rispettiva documentazione e garantendone la tracciabilità.»
L’importanza attuale del Rapporto ISTISAN 15/9 è confermata dal fatto che molti dei modelli organizzativi attuali propongono i workflows del suddetto Rapporto. Per esempio:
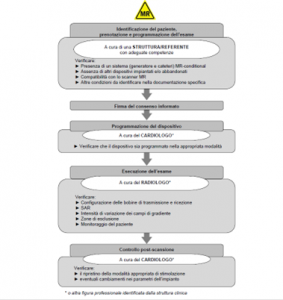
I modelli organizzativi specifici
A quest’ultimo proposito, il D.M. 14.1.2021 stabilisce i seguenti elementi minimi che deve possedere il modello organizzativo specifico:
- metodologia adottata per la identificazione univoca del dispositivo medico;
- procedura per l’identificazione della categoria di appartenenza del dispositivo medico in relazione ai possibili rischi di utilizzo in RM (dispositivo con etichettatura «safe», «conditional», «unsafe»);
- attribuzioni delle figure professionali coinvolte nel percorso di valutazione tecnica pre-esame;
- codifica degli accertamenti sul paziente in corso di esame RM;
- verifica di funzionalità post-esame RM del dispositivo medico impiantato, ove applicabile.
Ciascuna struttura, nei limiti di questi elementi minimi, potrà poi personalizzare il modello per rispondere alle proprie esigenze operative.
Ma attenzione: è essenziale che i modelli organizzativi siano
- periodicamente aggiornati, e
- conformi alle linee guida, in quanto
l’osservanza delle prassi interne, difformi rispetto alle linee guida accreditate e prive di basi scientifiche, è del tutto irrilevante al fine di escludere la responsabilità del medico. Quest’ultimo non può ignorare le linee guida, qualora appropriate in relazione alle condizioni specifiche del paziente
(Cass. Pen., Sez. IV, n. 39015 del 19 ottobre 2022).
“La prassi ospedaliera (può), al più, porsi come elemento che facilita o complica l’attività diagnostica del medico, ma che non può impedirla, né ritardarla, ove il medico facendo riferimento alle leges artis, si premuri di completare analisi e le valutazioni, che gli competono e che, in ogni caso, la struttura sanitaria assicura. E ciò anche se la prassi ospedaliera imponga una stretta e continuativa interlocuzione con gli altri reparti, non offrendo le agevolazioni logistiche che favorirebbero la semplificazione dei compiti.
È pur vero che la gestione del rischio clinico è definita da un intreccio di relazioni tecniche ed amministrative tra organizzazione sanitaria ed operatori, da un lato, e dalla diretta relazione terapeutica fra il paziente ed il medico, dall’altro, ma compete a quest’ultimo assicurare che il percorso diagnostico-terapeutico, comunque reso possibile dall’organizzazione sanitaria nella quale egli opera, sia portato efficacemente a compimento. Si tratta di obbligo che viene meno solo per cause di tipo logistico organizzativo, quale la sostituzione del medico che aveva originariamente svolto la visita di consulenza -e che attendeva l’esito degli esami da altri disposti al fine di completare la diagnosi- con altro collega, per motivi contingenti, come la fine del turno di lavoro, o l’impiego in altra attività urgente (…). In queste ipotesi, com’è ovvio, la responsabilità della definitività della diagnosi sarà posta a carico del medico subentrante”
(Cass. Pen., Sez. IV, n. 13573/2019). Trovate le due sentenze commentate qui).
Lavoro in équipe e ripartizione della responsabilità
In punto di lavoro in équipe e colpa professionale, il principio consolidato è quello per cui ogni sanitario è tenuto a vigilare sulla correttezza dell’attività altrui, se del caso ponendo rimedio ad errori che siano evidenti e non settoriali, rilevabili ed emendabili con l’ausilio delle comuni conoscenza scientifiche del professionista medio.
Tale principio, tuttavia,
«non opera in relazione alle fasi dell’intervento in cui i ruoli e i compiti di ciascun operatore sono nettamente distinti, dovendo trovare applicazione il diverso principio dell’affidamento, per cui può rispondere dell’errore o dell’omissione solo colui che abbia in quel momento la direzione dell’intervento o che abbia commesso un errore riferibile alla sua specifica competenza medica, non potendosi trasformare l’onere di vigilanza in un obbligo generalizzato di costante raccomandazione al rispetto delle regole cautelari e di invasione negli spazi di competenza altrui.»
(Cass. Pen., n. 36044 del 26 settembre 2022, commentata qui).
Per quello che ci consta, dunque, senz’altro non ci si aspetterà che l’aritmologo sia in grado di eseguire materialmente l’esame di risonanza magnetica, o che il radiologo provveda alla riprogrammazione del dispositivo impiantabile del paziente all’esito dell’esame: in queste fasi specifiche ed altamente specialistiche, ciascun operatore sanitario sarà unicamente responsabile nel caso di suoi errori.
Utilizzo off-label dei dispositivi
Infine, in tema di utilizzo off-label (cioè fuori protocollo) dei dispositivi medici, i requisiti di riferimento sono quelli indicati dall’art. 3, comma 2 della Legge 94/1998 (ex “Legge Di Bella”), in tema di farmaci, a mio avviso applicabile analogicamente anche in materia di dispositivi.
L’impiego off-label può essere adottato dal medico, sotto la sua responsabilità:
- “in singoli casi”, cioè in ipotesi specifiche ed individualmente definite, sulla base di criteri dettati dal beneficio che ci si potrebbe presumibilmente attendere per il paziente dall’esito dell’esecuzione dell’esame (rispetto alla sua non esecuzione)
- qualora ritenga, “in base a dati documentabili”, che il paziente non possa essere utilmente sottoposto a metodologie diagnostiche alternative e prive del margine di rischio presentato dalla RM
- “purché tale impiego sia noto e conforme a lavori apparsi su pubblicazioni scientifiche accreditate in campo internazionale”: maggiori sono gli scritti pubblicati, minore si ritiene essere il rischio al quale è esposto il paziente
- dopo aver documentato sia l’assenza di alternative utili e regolarmente registrate per quella indicazione (diagnostica), sia il razionale clinico dell’appropriatezza e della sicurezza dell’uso del dispositivo nel contesto clinico di utilizzo
- dopo aver fornito al paziente un’informazione completa ed effettiva sull’utilizzo scelto e sulle sue conseguenze
(in punto di consenso del paziente, vedi Cass. Civ., n. 18283 del 25 giugno 2021, commentata in “Utilizzo off-label del farmaco e responsabilità da omesso consenso del paziente“).
Ci aggiorniamo presto con un nuovo, interessante argomento!
Nel frattempo, resta collegato o iscriviti alla newsletter per non perdere i prossimi aggiornamenti.
A presto!
VEDI L’EVENTO
Aritmologia in Liguria: Core Curriculum per lo specialista clinico – Genova, 5 novembre 2022

